Analysis pipeline v3¶
Overview¶
Version 3 of the pipeline was released in June 2016 and includes the following updates and changes:
- Updated tools: InterProScan, FraGeneScan, QIIME and Trimmomatic
- Updated GO slim, based on the analysis of over 22 billion (22x10^9) billion functional annotations
- Added identification and masking of transfer RNA genes
- Improved quality control statistics (sequence length summary, GC and nucleotide distribution)
Figure 1 gives you a visual overview of the main steps and tools included in this version:
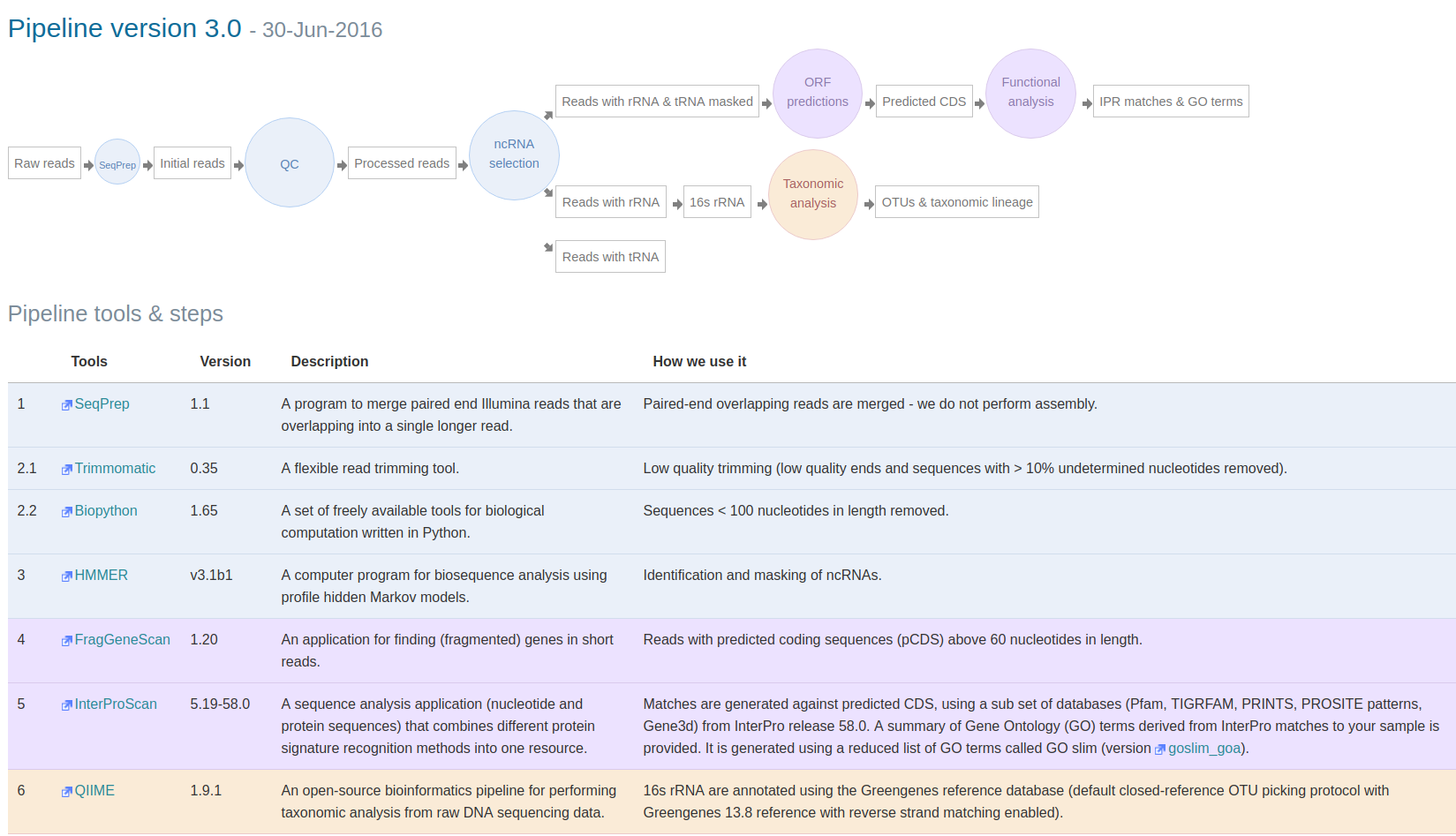
Figure 1. Overview of steps and tools included in pipeline v3:
Taxonomic analysis¶
EBI Metagenomics provides taxonomic analysis of sequences predicted to be 16S rRNAs using Qiime and the GreenGenes database for annotation.
The presence of 16S rRNA sequences in reads are predicted using HMMER and prokaryotic rRNA models from rRNASelector in forward and reverse orientation. The predicted 16S rRNA sequences are extracted from the reads and annotated using the Qiime closed-reference protocol for version 2 and higher of the pipeline. The taxonomic lineages are provided in at the kingdom, phylum, class, order, family, genus and species taxonomic levels (labelled k, p, c, o, f, g and s, respectively). Predicted 16S rRNA sequences that were not annotated by Qiime are grouped at the ‘Root’ level.
Other non-coding RNAs¶
For the identification of tRNAs within metagenomics datasets we are using HMMER and Rfam tRNA models.
Functional analysis¶
Functional analysis of predicted coding sequences (pCDS) from metagenomic data is provided using the InterProScan software provided by the InterPro database. InterPro is a sequence analysis resource that predicts protein family membership, along with the presence of important domains and sites. It does this by combining predictive models known as protein signatures from a number of different databases into a single searchable resource. InterPro curators manually integrate the different signatures, provide names and descriptive abstracts and, whenever possible, add Gene Ontology (GO) terms.
Protein signatures¶
Protein signatures are obtained by modelling the conservation of amino acids at specific positions within a group of related proteins (i.e., a protein family), or within the domains/sites shared by a group of proteins. InterPro’s different member databases use different computational methods to produce protein signatures, and they each have their own particular focus of interest: structural and/or functional domains, protein families, or protein features, such as active sites or binding sites (see below).
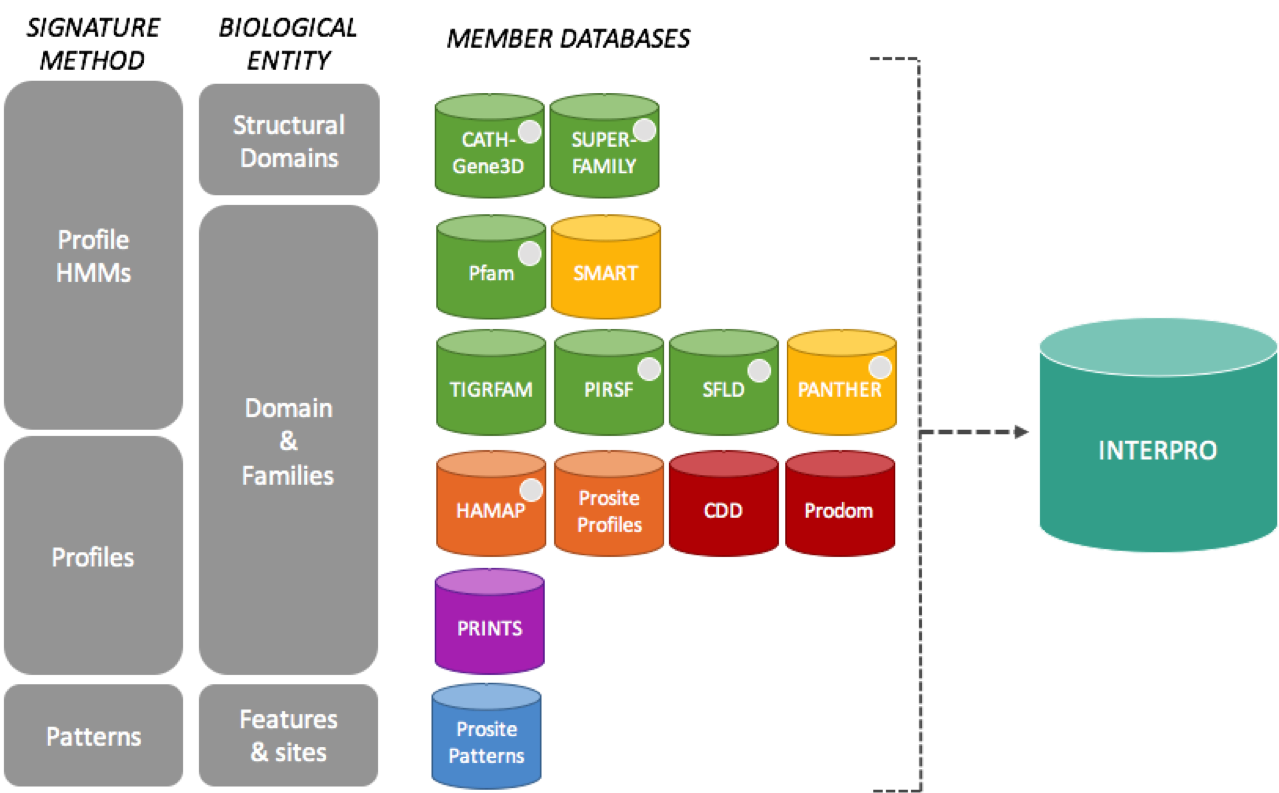
InterPro member databases grouped by the methods used to construct their signatures and focus of interest.
Only a subset of the InterPro member databases are used by EBI Metagenomics: Gene3D, TIGRFAMs, Pfam, PRINTS and PROSITE patterns. These databases were selected since, together, they provide both high coverage and offer detailed functional analysis, and have underlying algorithms that can cope with the vast amounts of fragmentary sequence data found in metagenomic datasets.
Assigning GO terms to metagenomic sequences¶
While InterPro matches to metagenomic sequence sets are informative in their own right, EBI Metagenomics offers an additional type of annotation in the form of Gene Ontology (GO) terms.
The GO is made up of 3 structured controlled vocabularies that describe gene products in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner. By using GO terms, scientists working on different species or using different databases can compare datasets, since they have a precisely defined name and meaning for a particular concept.
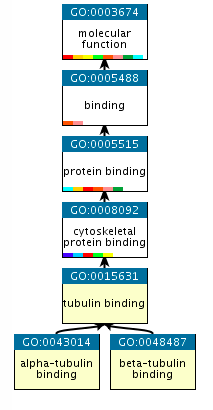
An example of GO terms organised into a hierarchy.
Terms in the GO are ordered into hierarchies, with less specific terms towards the top and more specific terms towards the bottom. (e.g., alpha-tubulin binding is a type of cytoskeletal binding, which is a type of protein binding). Note that a GO term can have more than one parent term. The Gene Ontology also allows for different types of relationships between terms (such as ‘has part of’ or ‘regulates’). The EMG analysis pipeline only uses the straightforward ‘is a’ relationships. More information about the GO can be found on the GO consortium documentation page.
As part of the metagenomic analysis pipeline, GO terms for molecular function, biological process and cellular component are assigned to pCDS in a sample by via the InterPro2GO mapping service. This works as follows: InterPro entries are given GO terms by curators if the terms can be accurately applied to all of the proteins matching that entry. Sequences searched against InterPro are then associated with GO terms by virtue of the entries they match - a protein that matches one InterPro entry with the GO term ‘kinase activity’ and another InterPro entry with the GO term ‘zinc ion binding’ will be annotated with both GO terms.